Hi-C技术源于染色体构象捕获技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。基于Hi-C数据中染色质片段间的交互强度呈现出随距离衰减的规律,Hi-C可以用于基因组组装,将杂乱的基因序列组装到染色体水平。
已有基因组组装水平提升
基因组de novo辅助组装
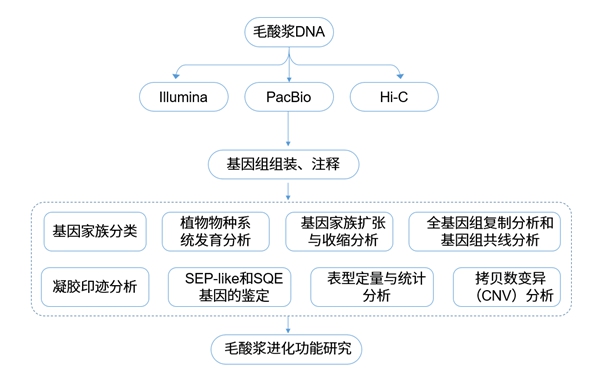
毛酸浆高质量参考基因组组装及其进化研究
The Physalis floridana genome provides insights into the biochemical and morphological evolution of Physalis fruits
期刊:Horticulture Research 合作方式:共同作者 影响因子:6.793 发表时间:2021.11
合作单位:中国科学院植物研究所 研究对象:毛酸浆
茄科是提供营养和果蔬多样性的重要来源之一。酸浆属作为茄科最大的属之一,具有独特的果实形态和生化多样性,是生态、进化和发育研究的新的园艺作物。毛酸浆作为酸浆属模式生物的代表物种之一,其花萼在受精后随浆果发育迅速膨大,形成“中国灯笼”或膨大花萼综合征(ICS)这一创新形态,包裹浆果。然而,酸浆属果实形态、生化特性及其起源背后的遗传基础还不够清楚。为了更好地了解茄科此类性状的起源、进化和发育机制,研究者使用PacBio三代测序技术和基于染色体构象捕获的Hi-C技术,实现了毛酸浆染色体水平的基因组组装。
材料选择:二倍体的毛酸浆幼叶
测序策略:
PacBio Sequel II 基因组测序,90X
lllumina DNA小片段文库,PE150双端测序,100X
lllumina Hiseq X Ten,Hi-C文库
结合进化和功能分析,对酸浆属果实形态创新“中国灯笼”和特征性化学成分的进化遗传基础有了新的认识。
高水平的毛酸浆基因组组装
首先,对基因组进行Illumina测序,共得到149 Gb的高质量数据。研究发现基因组纯合度较高,并且有大量的重复序列。通过K-mer分析后,利用PacBio Sequel平台进行测序,得到125Gb的subreads,组装成922条contigs,contig N50长度为4.87 Mb。并利用Hi-C数据将1.37 Gb的数据组装为12条染色体,使用Illumina二代测序数据验证,并与全长转录组数据进行比对,来评估组装的完整性,基因组组装评估表明得到了高水平的毛酸浆染色体水平基因组。

图1 毛酸浆的形态和基因组组装结果
基因组注释和基因预测
利用蛋白质编码基因、tRNA和rRNA基因、转座因子及GC分布和基因组重排事件等信息,进行数据评估。结合de novo和同源性的方法,将数据整合到毛酸浆基因组中,并在转录组序列辅助下,注释蛋白质编码序列。在当前基因组版本中,共预测出32,075个完整蛋白编码基因,进一步预测了3655个rRNA,997个tRNA,375个miRNA,和3047个snRNA 。

图2 高质量的毛酸浆基因组
基因组进化分析
利用7553个单拷贝基因家族进行系统基因组分析,揭示了毛酸浆和其他12个代表性物种的拓扑结构。考虑到茄科植物共同的WGD历史,进一步研究了酸浆属、番茄属和辣椒属三种基因组。在全染色体水平上,三个基因组之间存在明显的一对一共线关系,虽然基因组大小不同,但总体基因组是相对保守的,仅观察到几个小规模的染色体重排和物种特定的基因组区域。

图3 茄属植物的基因组进化
基因家族的进化分析
选取八种茄科植物的基因组,OrthoMCL法构建正交群,发现在茄科基因组中,大部分基因聚集在直系同源基因或旁系同源基因中;且某些基因家族在进化过程中经常发生扩张和收缩。在进一步揭示基因组独特性的研究中,选用毛酸浆和其他12种植物,发现直系同源基因、旁系同源基因和非聚类基因在13种植物中以相似的方式分布。GO富集分析中,毛酸浆特异的基因则发挥了多种作用。在以上多层次的进化分析中,在酸浆属、辣椒属和茄属植物的基因组中观察到小规模突变。其中,与甾体相关化合物生物合成途径关键步骤相关的基因和与“中国灯笼”形成相关的MADS-box基因的潜在遗传变异是关注的重点,并做了进一步研究。

图4 被子植物代表性物种的基因家族比较
参与类固醇化合物生物合成途径的基因进化
类固醇是所有真核生物必需物质之一,检测了类固醇在生物合成途径的关键基因。在33个基因家族中的结果研究表明,大多数基因家族的拷贝数变异(CNV)具有可比性,注释的HMGR和SQE基因的拷贝数发生显著变化,毛酸浆中发现20个SQE-like基因(拷贝数比非毛酸浆基因组高出约4倍),因此表现出特异性扩张。在甾体生物合成途径中,SQE基因编码黄酮类单加氧酶,在类固醇生物合成途径中起限速酶的作用。这些基因的CNV在与毛酸浆相关的不同物种间类固醇代谢产物的自然变异中所起的作用需要进一步深入研究。

图5 毛酸浆中参与类固醇相关衍生物合成基因的特异性进化
毛酸浆“中国灯笼”发育可能与 MBP21的缺失有关
毛酸浆基因组包含136个推定的MADS-box基因。研究者对MADS-box基因的进化关系进行了研究。排除基因组错误组装的可能性,数据同源性搜索结果证实毛酸浆和辣椒中MBP21发生缺失。这种缺失可能是进化过程中的染色体重排造成的。MBP21类基因的敲除或突变,往往产生花萼增大的现象。因此,这一基因丢失很可能与“中国灯笼”的起源有关。MPF2和MBP21基因的共同表达影响了酸浆属雄性不育和花萼的生长,表明酸浆属中MPF2和MBP21间的功能性冲突。这种功能冲突意味着这两个基因之间的协调是它们在ICS发育中正常发挥功能所必需的。


图6 酸浆属MBP21-like MADS-box基因丢失和“中国灯笼”的形成
茄科MBP21丢失与ICS出现的关系
不同物种基因组的Southern印迹分析结果表明,酸浆族中MBP21同源基因的丢失与ICS出现有密切关系。综上所述,MBP21类基因的丢失可能代表了一种与“中国灯笼”起源有关的新突变。

图7 酸浆族MBP21丢失与ICS的发生相关
研究者构建出第一个毛酸浆的高质量染色体水平的参考基因组。除了已知的MPF2的异位表达,基于基因组数据还发现一个SEP类MADS-box基因的丢失这一突变。它们可能共同决定了ICS(膨大花萼综合征)这一创新形态的起源。酸浆果实中某些甾类化合物大量积累可能与SQE基因起源和显著扩展有关。这些甾体的自然变异可能是基因家族的拷贝导致的。本研究强调了基因获得和基因缺失在果实新性状进化和发育中的重要作用,毛酸浆基因组数据为研究植物进化遗传学以及其他茄科作物的遗传改良和育种提供了重要参考。
Lu J, Luo M, Wang L, et al. The Physalis floridana genome provides insights into the biochemical and morphological evolution of Physalis fruits[J]. Horticulture Research, 2021, 8(1):244.


