

2023年9月16日,西北农林科技大学动物科技学院姜雨团队在Genome Research上发表题为“A Chinese indicine pangenome reveals a wealth of novel structural variants introgressed from other Bos species”的研究论文。该研究通过高质量中国黄牛泛基因组学分析,揭示了中国南方黄牛的驯化和演化机制。安诺优达为本研究提供三代PacBio HiFi测序服务。

01 研究背景
作为重要的现代饲养家畜之一,牛在人类文明发展的进程中对社会生产提升和人类文化进步做出了重要的贡献。家养牛主要分为起源于印度河流域的瘤牛(Bos indicus)和近东流域的普通牛(Bos taurus)。随着历史文明的发展,这些牛逐渐被传播,并分化为六个常见的牛群种类:欧洲普通牛、非洲普通牛、亚洲普通牛、印度瘤牛、非洲瘤牛和中国瘤牛。现代科学普遍认为中国瘤牛在3,500至2,500年前传入中国。随着群体的扩张,遗传多样性会丧失,但是中国瘤牛的多样性至少是其它牛群的两倍,且发现有部分来自野牛的遗传性质。因此,探究中国瘤牛中杂交遗传物质的数量、这些杂交对牛群遗传多样性的影响以及杂交畜群中的功能变异的贡献是有趣的科学问题。
02 研究结果
10个代表性中国黄牛品种的高质量基因组的从头组装
研究人员基于18-24X的测序深度,利用hifiasm完成20个基因组的初步组装,并使用RagTag完成挂载,获得10个高质量的染色体水平的参考基因组。组装完成的基因组大小在2679-2714 Mb之间,由110-367个contig组成,N50为18-91 Mb,平均BUSCO评估约95.5%。基于二代数据评估的基因组准确度QV值可达39.13%,碱基准确度大于99.99%。染色体完整性评估发现,组装基因组平均每个染色体包含962.18Kb的着丝粒序列和2.26Kb的端粒序列,高于已经发表的参考基因组。综上,研究人员构建了高质量的牛泛基因组图谱。
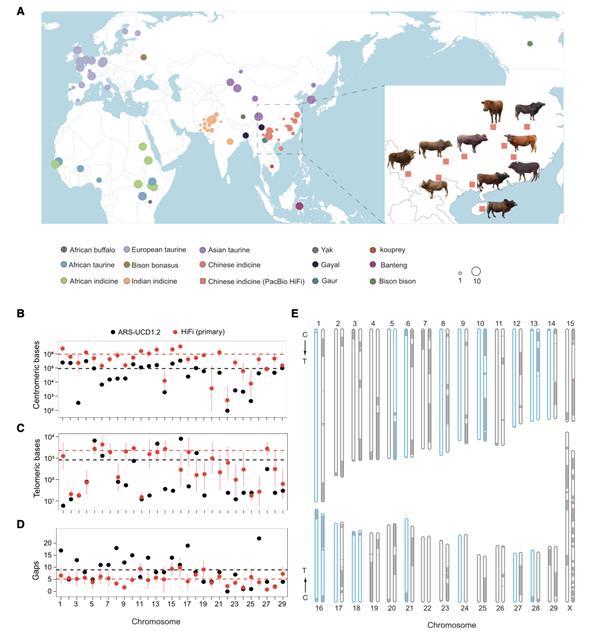
图1 不同品种牛的地区分布和高质量染色体水平基因组构建
构建中国黄牛的多重组装图谱
研究人员利用Hereford品种的线性参考基因组作为骨架,将之前组装的20个单体型参考基因组进行组装整合,构成多重组装图谱。在进行图形泛基因组的构建中,使用minigraph工具,并忽略了同源区域,对足够分散的子序列(长度大于50 bp)进行处理,创建新节点("bubbles")。多重组装图谱中包含了160,000个非参考节点,总跨度为148.5 Mb,其中包括约22.21%的泛基因组(即不被所有组装基因组共享的灵活序列)。从图形泛基因组中提取了74,907个非参考等位基因(长度大于100 bp),并与牛属(Bos species)的整个基因组的重测序数据进行比较,发现有53.96%的新序列在其他瘤牛/普通牛种群中无法检测到,其中包括26.21%在野生牛属物种中特有的序列。
此外,研究人员发现了丰富的重复变异,其中LINE/L1占主导地位,多等位性变异中以可变数串联重复(VNTR)为主要类型。研究人员使用de novo和同源性的方法,进行新基因和基因结构的预测。结果表明,预测到1,153个完整的基因模型,其中有271个基因是新发现的基因,这些基因家族可能在嗅觉转导、免疫应答、信号传导、内源性逆转录病毒-K蛋白质以及核糖体蛋白等多个生物过程中发挥作用。
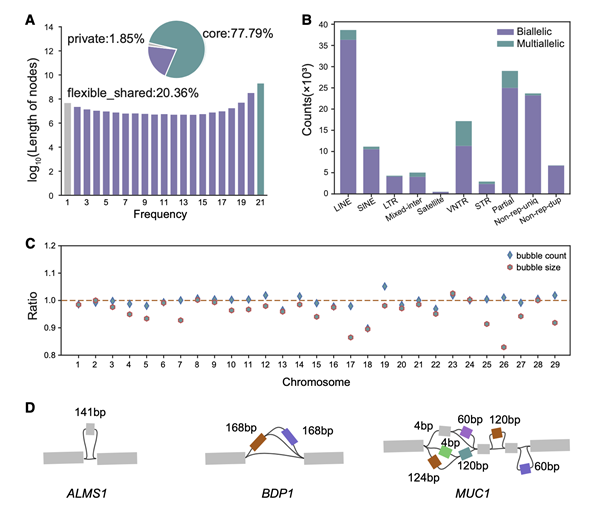
图2 中国黄牛的多重组装图谱
中国黄牛结构变异的生成和特征分析
研究人员使用多种SV检测工具,建立了中国黄牛的结构变异(SV)目录,并对SV进行了详细的分类和特征化。具体而言:通过合并从所有样本中检测到的高置信度SVs,构建了一个包含156,000个非冗余的长度大于等于50 bp 的SVs目录,其中包括73,889个删除和82,120个插入。这些SVs覆盖基因间区和内含子区域,且包括一小部分的外显子重叠。接着,将中国黄牛的非冗余SV目录分为四个类别:共享(在所有样本中都被发现的)、主要(在至少50%的样本中被发现,但不是所有样本)、多态(在1%至50%的样本中被发现)、单一(只在一个样本中被发现)。
SV分布分析表明,大多数SV相对较短,其中包括两个长度约为145 bp和285 bp的峰值,主要注释为BOV-A2(SINEs),一个长度为1,295 bp的峰值对应ERV2-LTR-BT,而一个长度为8,500 bp的峰值对应LINE/L1,这表明转座元件是牛中SV的重要来源。SV热点分析鉴定了206个SV热点,跨越了约195 Mb的基因组,其中61个位于染色体末端的最后5 Mb内。28个热点与先前发布的已鉴定到的热点重叠,而其余119个热点是新的,此外,对SV热点中检测到2,533个蛋白质编码基因的功能预测表明,这些基因主要与免疫系统和嗅觉传导相关。
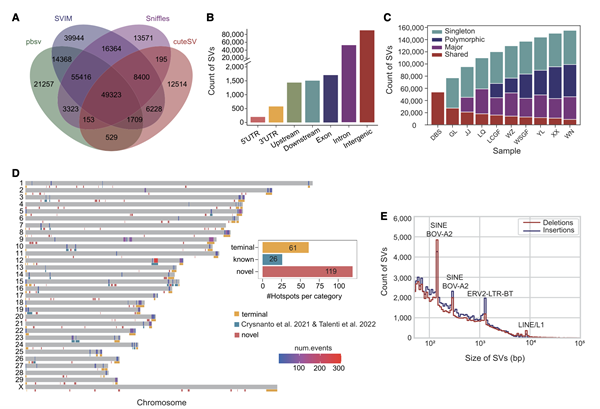
图3 10个中国黄牛结果变异的特征分析
不同牛品种对中国牛的遗传贡献
研究人员使用隐马尔可夫模型,分析20个单体型的中国黄牛基因组,并将其与321个非中国黄牛的基因组进行比较。研究发现,中国黄牛的基因组中包含来自多个牛属物种的古代引入的基因片段。不仅有来自野牛(banteng)的基因片段,还有另外的来源,如古代的林牛。
这些引入的片段涵盖了中国黄牛基因组的大部分,占总基因组的73.3%;相比于常染色体,X染色体中的古代引入元素明显较少,这可能是由于更强的自然选择或对不兼容外源等位基因的敏感性所致;对于遗传多样性,研究发现,引入的基因片段使中国黄牛的基因组具有更高的核苷酸多样性(θπ),而在引入区域的变异数量也明显高于非引入区域,这些观察结果表明,引入的基因片段对中国黄牛的遗传多样性产生了积极影响;此外,研究估计了不同来源物种的基因引入发生的时间,结果显示不同来源的基因引入发生在不同的时间和地点,其中野牛样来源的基因引入发生时间最早,在约3,360-3,192年前。研究还发现,野牛样和林牛样基因片段的全局含量与海拔呈显著负相关,表明不同牛群的栖息地可能导致了不同的遗传结构或后续选择压力。
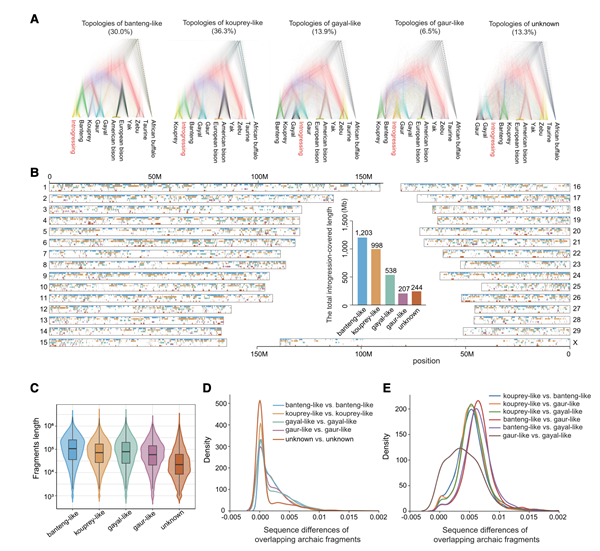
图4 中国黄牛的基因渗入和演化分析
03 研究结论
研究人员使用了来自中国南部的10头中国黄牛的PacBio HiFi测序数据,组装了20个高质量的单体型基因组,并将它们整合到一个多组装图中,其中包含了148.5 Mb(5.6%)的新序列。研究还鉴定了156,009个高置信度的非冗余结构变异(SVs)和206个SV热点。此外,研究检测到34,249个古代遗传片段,覆盖了中国黄牛基因组的73.3%,这些古代遗传片段的来源包括了来自爪哇牛、林牛、大额牛、印度野牛等不同牛属物种的遗传物质,以及0.6%的未知来源的遗传物质,多个不同的物种的遗传物质可能为中国黄牛的遗传多样性做出了贡献。总之,这项研究强调了不同物种间的遗传物质迁移对于一个重要的家畜群体的基因组结构所起到的作用,并展示了异源基因组元素如何为选择提供了可用的遗传变异,对于动物进化研究和遗传育种具有重要的参考意义。
参考文献
Dai X, Bian P, Hu D, Luo F, Huang Y, Jiao S, Wang X, Gong M, Li R, Cai Y, Wen J, Yang Q, Deng W, Nanaei HA, Wang Y, Wang F, Zhang Z, Rosen BD, Heller R, Jiang Y. A Chinese indicine pangenome reveals a wealth of novel structural variants introgressed from other Bos species. Genome Res. 2023 Sep 15. doi: 10.1101/gr.277481.122. Epub ahead of print. PMID: 37714713.