
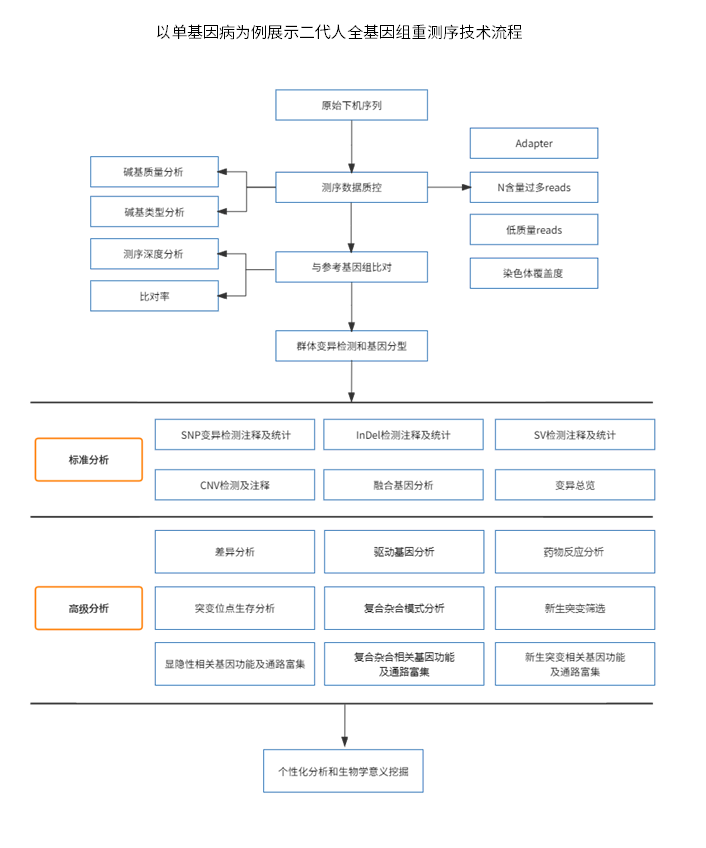
二代人全基因组重测序 (Whole-genome sequencing,WGS)是基于二代测序数据分析基因组的方法。可以同时研究编码区和非编码区的DNA变异与疾病之间的关联。
产品优势
- 项目经验项目合作 1300+
- 合作文章累计影响因子 260+
- 项目团队1V1 医学项目服务
- 医学多组学分析流程涵盖基因组、医学转录组及表观组等组学数据联合分析
- 关注临床医学前沿的研究成果针对癌症/疾病的医学报告
- 临床转化经验安诺血液病临床转化产品
应用领域
- 单基因病
- 复杂疾病
- 肿瘤研究
- 群体研究

二代人全基因组重测序 (Whole-genome sequencing,WGS)是基于二代测序数据分析基因组的方法。可以同时研究编码区和非编码区的DNA变异与疾病之间的关联。
| 样本类型 | 起始量 | 具体操作 |
| 血 液 | 1 mL | EDTA抗凝管采血,颠倒混匀,冻存管分装 液氮速冻,-80 °C保存,干冰运输 |
| 新鲜组织 | 100 mg | 迅速取材,离体后立即用0.9%的生理盐水清洗 分割为2-3 mm的组织块,冻存管保存 液氮速冻,-80 °C保存,干冰运输 |
| 人体FFPE | 白片8片 石蜡卷8卷 | 厚度 5-10μm,表面积 10mm*10mm 常温保存及运输,注意避免不同样品间蜡片交叉污染 建议使用1年内的FFPE样本 |
| 细 胞 | 106-107个 | 悬浮细胞:去除培养液,PBS清洗2遍 贴壁细胞:去除培养液,胰酶消化,PBS清洗2遍 -80 °C保存,干冰运输 |

本研究对39个CIS病变进行全基因组测序,并与来自癌症基因组图谱(TCGA)的肺鳞癌(LUSC)外显子序列数据进行比较,发现CIS标本和TCGA LUSC肿瘤之间有相似的突变负荷和拷贝数分布。两个数据集之间没有统计上的显著差异,没有一个单一的癌症突变可以完全区分进展型和消退型病变。
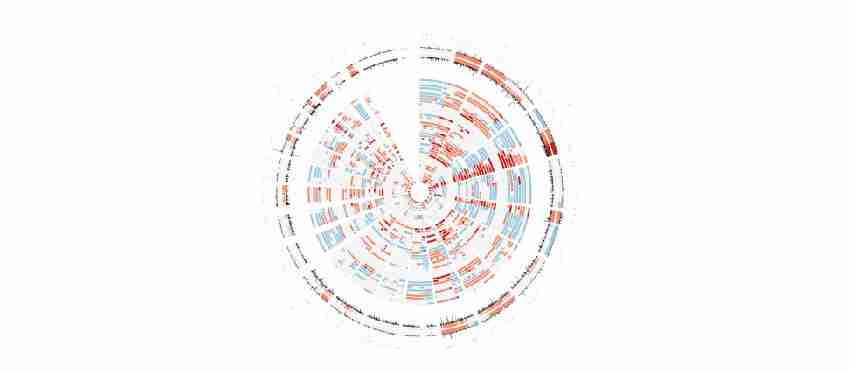
图1 肺癌原位病变(CIS)基因组与TCGA中肺癌数据比较Circos图
对17个进展型和16个消退型CIS病变进行检测,发现有1,335个基因有明显的表达变化。在进展型CIS病变中,657个基因表达上调,678个基因表达下调。进一步结合26个进展型、11个消退型和23个对照样本的甲基化图谱差异分析结果,发现Nkx2-1(TTF-1)是唯一被鉴定的候选驱动基因,在进展型样本中呈高甲基化和低表达。对基因表达和甲基化数据的主成分分析显示,进展和消退亚组之间有明显的区别。
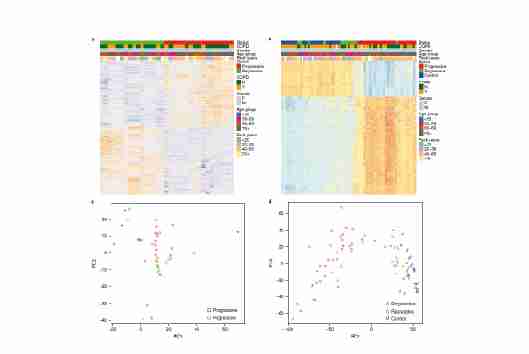
图2 原肺原位病变中进展性与退行性甲基化和基因表达的差异
为预测侵袭前病变是否会进一步发展为癌症,本研究建立模型对基因表达进行分析。当应用于TCGA的数据时,该基因模型能够对LUSC和对照样本进行分类(图3a-c)。对甲基化进行类似的分析,观察到与TCGA对照组相比,TCGA LUSC中等水平甲基化的探针数量有所增加(图3d),揭示样本间存在甲基化异质性。
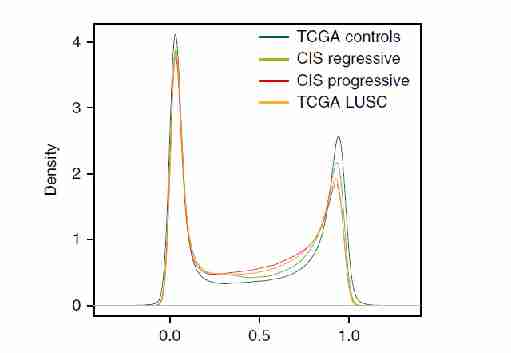
图3 Methylation beta-value 甲基化图谱预测癌症的进展
该研究通过多组学数据分析癌前病变CIS的遗传学特征,构建可以区分肺鳞癌组织与正常组织的转录组与甲基化模型,同时评估其对进展型与消退型CIS进行预测的可行性,并发现NEK2基因高表达极有可能预示所在病变组织会发展为肺鳞癌。该研究发现的生物标志物有助于开发检测早期癌症发展相关分子信号的方法,对肺鳞癌早期病变的筛查与预防具有指导意义,可能使更多病人受益。
Teixeira V H , Pipinikas C P , Pennycuick A , et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions[J]. Nature Medicine, 2019.